2011年6月美国国家科学技术政策办公室组织了一个材料基因组计划(MGI),将材料计算设计、数据处理以及综合的工业化过程列为政府重点投资的方向,我国也特别组织了“材料科学系统工程”香山会议对材料计算设计进行了研讨。因为现代材料计算学的发展,通过材料计算而实现先进材料的设计成为可能。如果利用材料基因组的方法来进行材料设计,周期可以缩小一半,而费用则只有几分之一或几十分之一。 微观模拟和跨尺度模拟是材料基因组的核心组成。材料的性质,特别是力学性质,通常是多尺度的过程相互关联,包括从原子尺度到宏观尺度。目前我们的研究主要集中在基于第一性原理结果的原子势场开发,用反演势发展适合于跨尺度模拟的经验势场,并将其运用于TiAl合金等航空关键材料、锂电等储能材料的性质计算。课题组已经自主发展了基于Chen-Mobius反演的MEAM分子动力学程序,并且利用计算模拟成功预测了一些复合碳基材料的新构型和新型能。进一步,我们将设计低维碳基材料的杂化结构,包括纳米树芽、杂化管束、纳米支撑结构等,从理论上探索它们表现出来的新奇的力学、电学、光学等特性,并深入研究这些新型碳基杂化结构在纳米电子学以及能源材料如储氢,锂离子电池电极材料等方面的应用。
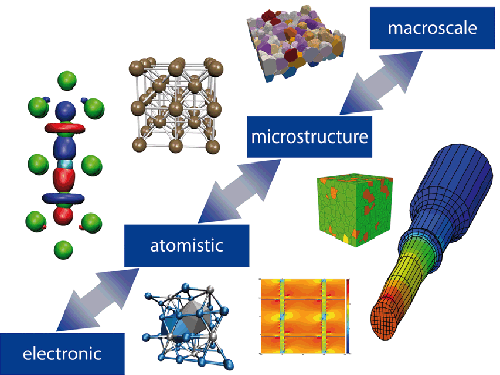
经过上世纪几十年的发展,一整套材料计算的方法已基本提出。而随着最近十几年内计算机性能的飞速提升,计算材料学的应用领域得到很大的拓展,模型复杂度也得到很大提升。一般来说,根据所研究体系的时间和空间尺度,可以将计算材料学分为第一性原理、原子模拟(分子动力学和蒙特卡洛法)、有限元或有限差分法等。
势场开发
原子模拟方法(分子动力学和蒙特卡洛法)是建立在牛顿力学的基础之上。它忽略了原子内部的复杂架构,而将其看作孤立的实心球体。在考虑原子之间的相互作用时,引入了原子间相互作用势的概念。原子间作用力的大小由势函数精确描述,而原子的受力演化过程则有牛顿运动定律描述。由于原子模拟理论简单,因此能够研究的体系很大。目前报道最大的研究体系达到了数以亿计的原子。另一方面,原子模拟能够研究的物理问题也很多,而且能够给出相对精确的结果,因此得到了非常广泛的应用。由于原子模拟的技术手段相对成熟,所以其模拟结果的准确性一般由原子间势函数单一决定。可以认为模拟过程中所采用的势函数是否准确描述实际原子间的相互作用,将直接影响到模拟结果的准确性。因此,采用原子模拟方法进行材料学研究的首要和核心问题,就是寻找可以精确描述原子间相互作用的势函数。
目前比较精确的原子间相互作用势是MEAM,然而由于只考虑了第一近邻原子的贡献,导致在描述一些BCC过渡族金属的基态和表面能时出现了问题。另外,MEAM考虑了多体屏蔽函数,它使得MEAM的复杂度大大增加。基于MEAM存在的这些问题,我们一方面移除了MEAM中多体屏蔽函数,另一方面通过使用考虑陈难先院士提出的Chen-Möbius晶格反演法,考虑了更多近邻原子的相互作用,提出了基于晶格反演法的MEAM势函数(LI-MEAM)。更进一步,我们将LI-MEAM势函数应用到了更多的体系中,构建了众多体系基于LI-MEAM的势库。
介观模拟
对高聚物的流变属性进行计算模拟,最直接的挑战在于模拟的长度尺度难于把握。高聚物分子量很大,如果在微观尺度上,如使用分子动力学进行模拟,将需要不可想象的计算时间;如果在宏观尺度上,如使用有限元的流体动力学方法进行模拟,又很难观测流变细节。
研究发现,在介观尺度上对许多高聚物的流变属性进行模拟,能够与实际的物理性质很好的吻合。1993年,Ladd在晶格波尔兹曼方法的基础上,考虑有限粒子碰撞的动量的传递,从而使这种内在的粒子间的相互作用能够很好地描述宏观粘性流体的流动。晶格波尔兹曼方法克服了传统流体动力学的许多不足,如直接将动能方程离散化以易于数值求解,将复杂的模型直接映射到三维的网格,大幅提高了计算效率。同时期的另外一种广泛应用到高聚物研究中的介观方法是布朗动力学。1991年Rudisill和Cummings发表了关于将布朗动力学应用于稀释的剪切聚合物流体的论文。这种方法用随机力来替代溶剂分子,因为快速运动的溶剂分子与缓慢运动的高聚物在时间尺度上相差很大。这就使得,布朗动力学能够进行比分子动力学更大时间尺度的模拟。这种方法非常适合流动力作用下的复杂流体结构和流变的研究。耗散粒子动力学最初是在1992年由Hoogerbrugge和Koelman在结合分子动力学和气体格子法的优点的基础上提出。这种方法可以自适应多种边界条件,并且不需要对区域进行网格化。它更像分子动力学,只不过实在其上进行了粗粒化,从而能够在介观尺度上进行模拟。耗散粒子动力学主要征对介观的复杂流体系统,主体框架使用的是分子动力学,只是对系统的建模过程和力学描述进行了规定。介观是相对微观和宏观来说的,一般从几十纳米到几微米。在分子动力学中,一个系统由粒子与粒子之间的相互作用势描述。耗散粒子动力学与分子动力学很类似,不过,在耗散粒子动力学中,每个粒子代表了多个原子或分子,这种处理方法称为粗粒化。粗粒化的程度需要根据实际仿真的体系来决定,粗粒化程度太低,模拟就需要很长的模拟时间;粗粒化程度太高,有可能就会丢失很多细节,得不到满意的结果。与晶格波尔兹曼模型不同的是,耗散粒子动力学模拟中的粒子可以随意的移动,而不是限定在特定的格点上,省去了进行网格化的麻烦的步骤。整个模型也因此变得简洁,便于实现。相对于布朗动力学,耗散粒子动力学可以显现地表示溶剂,从而易于进行高聚物与溶剂的相互作用的模拟,也可以通过高聚物与溶质之间的热力学描述反映体斥效应。
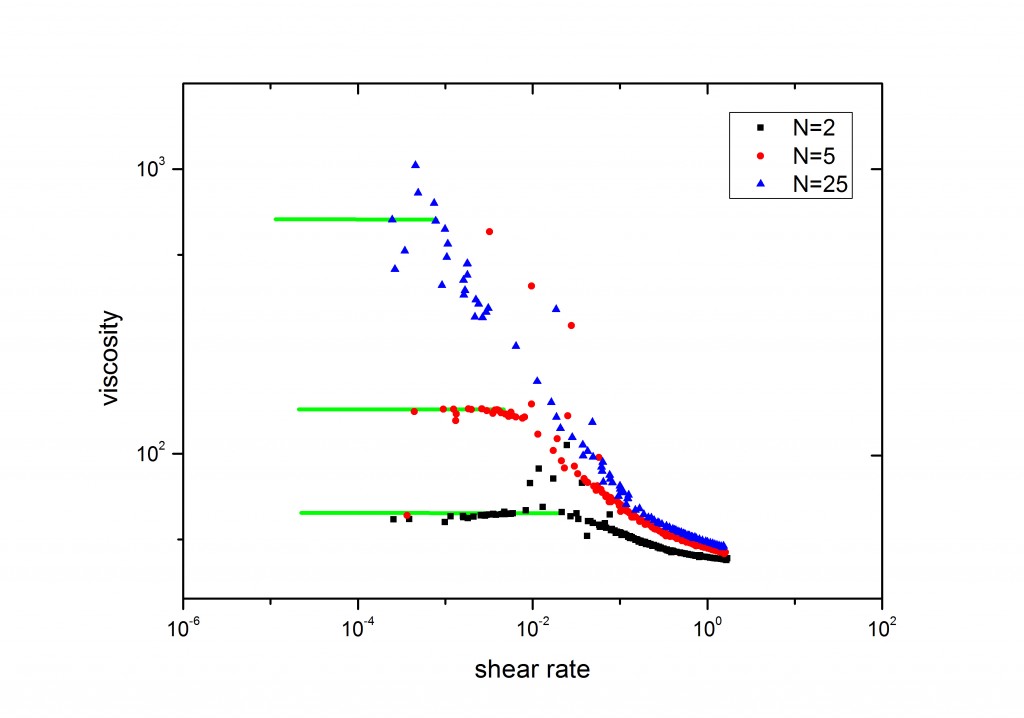
使用DPD计算聚合物的粘度属性