有机配体的表面修饰是显著提升钙钛矿纳米晶体稳定性的关键技术。然而,配体的筛选与设计过程往往依赖于研究人员的专业经验和直觉判断,并且需要通过耗时的实验来进行验证。鉴于候选化合物的化学空间极为庞大,传统的实验方法在效率上显得力不从心,迫切需要开发一种高效、大规模且成本低廉的策略来设计潜在的表面配体。在这一背景下,华中科技大学微纳中心的单斌教授、文艳伟副教授领导的研究团队在《Journal of Physical Chemistry C》上发表了他们的最新研究成果,这项研究基于第一性原理计算和主动学习框架,为CsPbBr3纳米晶体表面配体的设计提供了新的策略。
在这项工作中,研究团队利用了开源数据库ZINC,从中提取了超过16万个有机分子的SMILES(简化分子输入行项系统)表达式,并选定了六种分子描述符来构建特征空间。这些描述符的选取旨在综合评估分子与钙钛矿表面的结合强度以及分子在非极性溶剂中的溶解性,两者都是评价配体适用性的重要指标。通过这种基于计算的主动学习方法,研究团队能够高效地筛选出与CsPbBr3纳米晶体表面结合紧密、且在制备过程中具有良好溶解性的优质有机配体,大大加速了钙钛矿纳米晶体稳定性研究的进展。此外,这种方法的成功应用,不仅展示了计算化学和机器学习在高通量材料筛选中的巨大潜力,也为未来钙钛矿及其他功能材料的表面修饰设计提供了一条高效、经济的新途径。
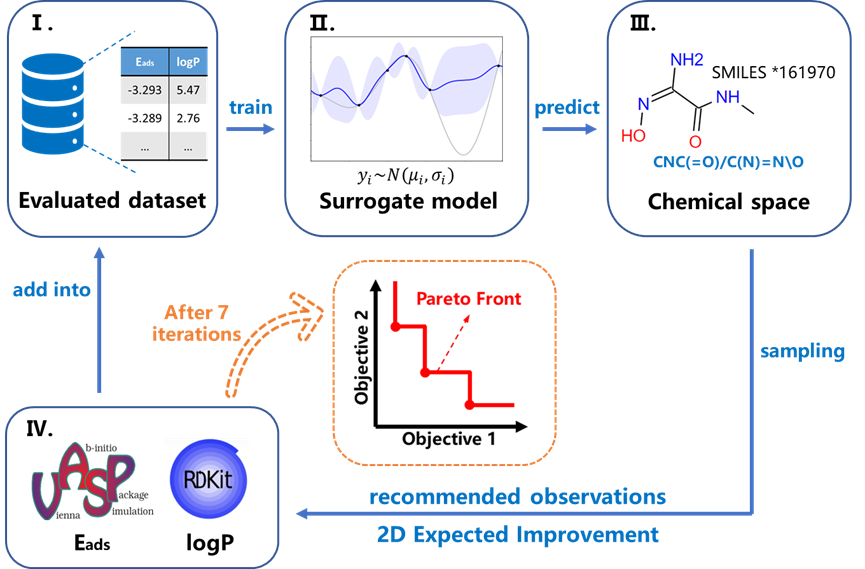
图1第一性原理计算辅助的主动学习框架示意图。

图2主动学习流程迭代产生的所有数据点。
该团队开发了一个以高斯过程回归作为代理模型的主动学习框架,采用了基于双目标期望改善函数和聚类方法的批量采样策略。通过第一性原理计算确定分子的吸附能,并根据分子结构估算脂水分配系数,从而全面评估一个分子作为表面配体的潜力。研究从一组包含典型官能团的简单有机分子出发,利用最远点采样策略在特征空间中补选分子,形成了初始数据集,并进行了7轮迭代学习。最终得到的帕累托最优解集包括了6个有机分子,这些分子提供了在两个关键目标量间不同权衡比重下的最优解案例。
此外,通过对候选分子的吸附构型和界面电子态密度等进行深入分析,确认了这些分子的键合模式,并验证了这些分子不会在纳米晶体的带隙中引入孤立的陷阱态,从而在稳定地结合到纳米晶表面的同时,不会对其荧光效率产生负面影响。在帕累托前沿附近的分子子集中,观察到了卤素、酮、亚胺、硫醚和苯环等子结构的集中出现,与文献中报道的优秀配体特征一致。
总而言之,该团队结合密度泛函理论和主动学习技术,开发了一套纳米晶表面配体快速筛选框架。通过仅80次的第一性原理计算,在超过16万个有机分子的庞大化学空间中,经过有限的迭代步骤,快速锁定了6个具有高预测精度的候选分子。微纳中心的王昭杰博士是论文第一作者,这项研究展示了数据驱动方法在加速材料设计中的巨大潜力。